维多利亚vic309官网程俊教授和杨勇教授研究团队合作,发展了基于机器学习计算顺磁电池材料动态NMR化学位移的方法,应用于P2型钠离子电池正极材料并进行实验验证。该工作以“A Machine Learning Protocol for Revealing Ion Transport Mechanisms from Dynamic NMR Shifts in Paramagnetic Battery Materials” 为题,发表在Chemical Science(2022, DOI:10.1039/D2SC01306A)。
固体核磁共振(solid-state Nuclear Magnetic Resonance, ssNMR)技术凭借其对局域环境和动态信息的独特敏感性,在电池材料的研究中得到了广泛应用。不过电池正极材料通常具有顺磁性,过渡金属离子与被观测核存在复杂的相互作用,可靠的NMR谱峰指认非常依赖密度泛函理论(Density Functional Theory, DFT)计算。已有理论方法被开发用于计算正极材料NMR化学位移,但这些方法仅适用于碱金属离子扩散缓慢的体系,其NMR谱峰与碱金属离子局域环境一一对应。而在高倍率电池正极材料中,例如P2型钠电正极,因碱金属离子在不同局域位点的快速扩散,常温下往往观测到动态NMR谱,其谱峰对应于多个碱金属离子局域环境的动态平均,这进一步加大了NMR谱峰的指认难度。
近年来,程俊教授和杨勇教授研究团队展望了分子动力学(Molecular Dynamics, MD)模拟与NMR在固体电池材料中的联合应用(Curr. Opin. Electrochem., 2022, 35, 101048.),并通过联用DFT计算局域位点NMR化学位移和深度势能分子动力学(Deep Potential Molecular Dynamics, DPMD)模拟,首次直接计算P2型Na2/3(Mg1/3Mn2/3)O2的动态23Na化学位移(Angew. Chemie Int. Ed., 2021, 60, 12547–12553.)。不过该工作中应用优化结构(0 K)来计算局域位点化学位移,忽略了热力学涨落对化学位移的影响,限制了其广泛应用。
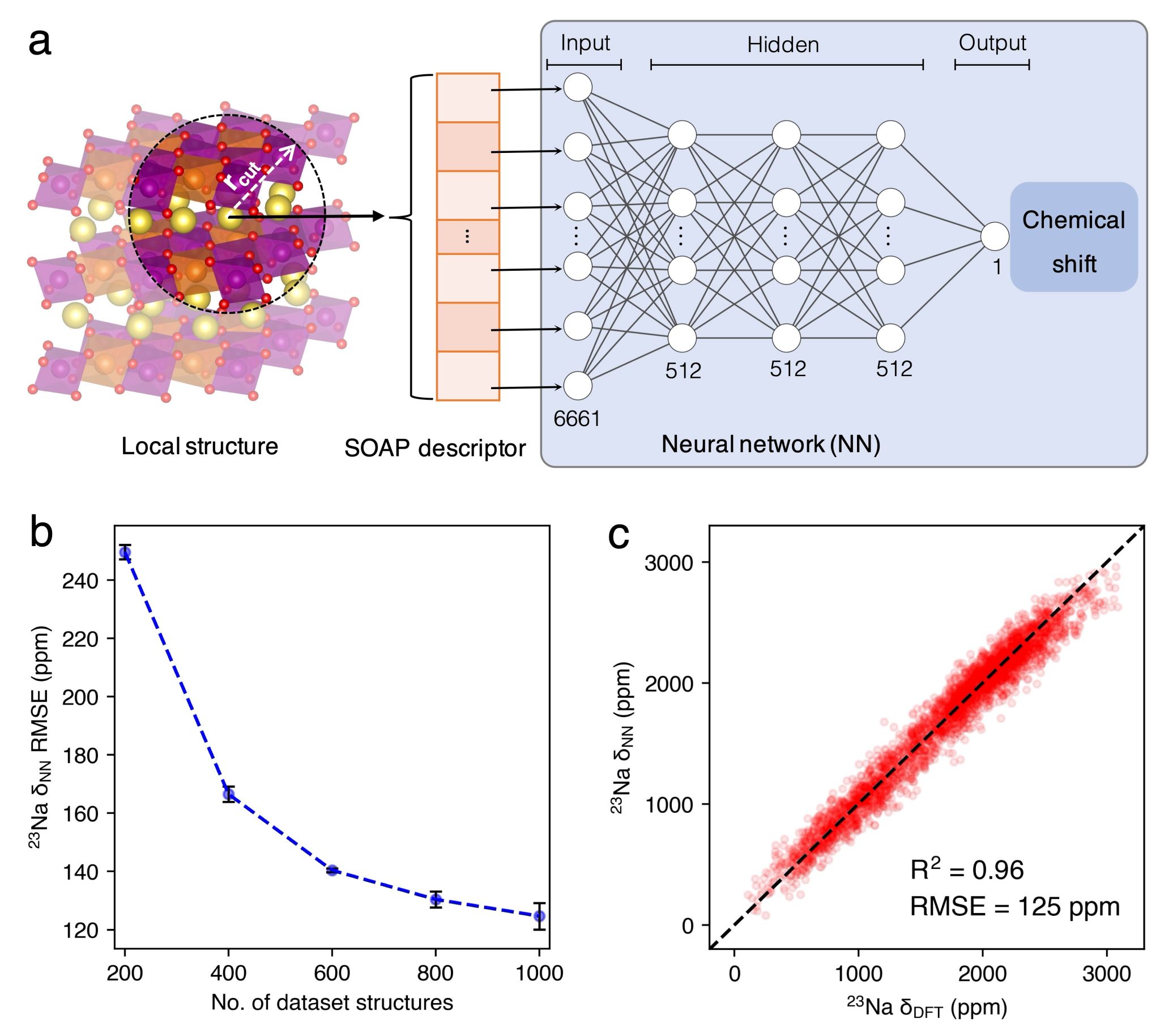
针对以上问题,本工作中通过联合DPMD模拟进行快速结构采样,和机器学习方法快速预测结构的化学位移,可在DFT精度下计算动态NMR化学位移。具体的,建立了一个基于DFT数据集,结合原子位置平滑重叠描述符(Smooth Overlap of Atomic Positions, SOAP)和神经网络(Neural Network, NN)的机器学习模型,名为NN-NMR模型。以P2型Na2/3(Mg1/3Mn2/3)O2为例验证了该模型,结果表明NN-NMR模型可以较好的复现DFT计算结果(R2 = 0.96)。此外,NN-NMR模型在GPU卡上计算时间与DFT方法在CPU核上计算时间相比,缩短了几个数量级。联合NN-NMR和DP两个模型的高效率和精度,该方法基本消除了P2型Na2/3(Mg1/3Mn2/3)O2和Na2/3(Ni1/3Mn2/3)O2动态23Na化学位移计算值的统计误差,并与实验测量结果一致。该方法可用于建立高倍率电池正极材料结构,NMR谱,离子传输之间的明确关系,且有潜力推广到其他的原子核,固态电解质,溶液体系中,作为联系实验NMR谱和微观动态过程的桥梁。
论文第一作者为维多利亚vic309官网2018级博士研究生林敏,该工作得到了美国国家强磁场实验室傅日强教授的支持和帮助。研究工作得到了国家重点研发计划(2021YFB2401800),国家自然科学基金(21991151、2199115、21935009、21761132030、21861132015、22021001、92161113、91945301)和厦门市科技计划项目(3502Z20203027)的支持。
论文链接:https://pubs.rsc.org/en/content/articlelanding/2022/SC/D2SC01306A

